Глава
12 КОРРОЗИЯ
Назад: 11.2 Характеристика
свариваемости и рекомендации по сварке.
12.1
Основные понятия и виды коррозии
Коррозией называется разрушение металлов и сплавов в
результате химического или электрохимического воздействия на их поверхность
внешней среды. В основе процессов коррозии металлов и сплавов лежит явление
окисления. Образование окислов металлов в большинстве случаев для земных
условий является термодинамически естественным, так как соответствует
снижению потенциальной энергии системы металл- среда. Именно поэтому
большинство металлов в природе существует в виде руд, т.е. окислов и на их
восстановление требуются затраты энергии.
Коррозия ведет к прямым и косвенным
потерям при эксплуатации деталей и сооружений, в том числе сварных. К прямым потерям относятся: стоимость
потерянного металла, изготовленных узлов и машин, снижение срока службы изделий
и затраты на противокоррозионные мероприятия. Косвенные потери связаны с временным выходом оборудования и сооружений
из строя или их простоями, получением некачественной продукции, увеличением
расходов на ремонт, ущерб окружающей среде и т.д.
Интенсивность взаимодействия металла или
сплава с коррозионной средой зависит от свойств самого металла и окружающей
среды. Сварка как технологический процесс, всегда вызывает локальное изменение
свойств исходного металла в определенной зоне и, следовательно, эти зоны будут
обладать иным характером взаимодействия металла со средой эксплуатации.
Химическая
коррозия – это взаимодействие
металла со средой, при котором окисление металла и восстановление
окислительной компоненты среды протекают в одном акте. При этом во
взаимодействующей системе не возникает электрического тока, а происходит
обмен электронами между металлом и окислителем. К таким видам взаимодействия
металла и среды относятся его реакции с кислородом, хлоридами, галогенами, с
кислородсодержащими газами при отсутствии влаги. Например:
Fe + 0,5О2 ![]() FеO.
FеO.
В приведенном примере железо Fe отдает
свои 2 электрона, превращаясь в положительный ион Fe++, а атом
кислорода получает 2 электрона, превращаясь в отрицательный ион О- -. Образовавшаяся система уравновешена и обладает
минимумом потенциальной анергии. В общем виде подобные реакции можно записать:
Ме + Х ![]() МеХ.
МеХ.
По условиям протекания процесса окисления
различают следующие виды химической коррозии:
1) газовая коррозия – коррозия металлов и
сплавов, вызываемая действием газов или воздуха при высоких температурах;
2) коррозия в неэлектролитах, т.е. в
жидкостях, не проводящих электрический ток (сернистая нефть, бензин, мазут,
спирты) при нормальных и повышенных температурах.
Продукт взаимодействия кислорода с
металлом – оксид образует на поверхности металла пленку, которая снижает
химическую активность металла и тормозит процесс дальнейшего окисления. В
зависимости от толщины оксидные пленки подразделяются на тонкие (невидимые),
толщиной до 40 нм; средние (видимые как цвета побежалости) – 40...500 нм;
толстые – более 500 нм.
Оксидные пленки могут быть сплошными и
несплошными. Условие сплошности выражается соотношением молекулярных объемов Vокс/
VМе > I. При Vокс/
VМе < I оксид является пористым и не защищает металл от
дальнейшего окисления. Скорость образования окисных пленок определяется взаимной
двусторонней диффузией агрессивной среды в металл и металла в среду. На
скорость газовой коррозии влияют температура, состав газовой среды и состояние
поверхности металлов. Температура существенно увеличивает скорость коррозии,
что видно из соотношения:
![]()
где Vкор – скорость коррозии; А, В –
константы; е – основание натуральных логарифмов; Т – абсолютная температура.
Для защиты от газовой коррозии используют
главным образом жаростойкие сплавы, основными легирующими в которых чаще всего
являются хром, алюминий и кремний. Например, введение в железоуглеродистый
сплав 12 % Cr делает его пассивным к кислороду не только при нормальных
температурах, но и при нагревании до T ![]() 700
700
Разновидностью химической коррозии
является коррозия металлов в
неэлектролитах, где активными реагентами являются сера, сероводород,
сероуглерод и др. Основными мерами борьбы против коррозии в неэлектролитах
является использование коррозионностойких (нержавеющих) или алитированных
сталей.
Электрохимическая
коррозия – это процесс самопроизвольного
разрушения металлов и сплавов при воздействии на них электролитов. К последним
относятся растворы кислот, щелочей, солей, техническая вода. Известно, что
молекулы электролита при растворении в воде диссоциируют, т.е. распадаются на положительно
заряженные ионы – катионы и отрицательно
заряженные – анионы (соль NaCl ![]() Na++Cl–; кислота НNО3
Na++Cl–; кислота НNО3 ![]() H++NO3–;
щелочь NaOH
H++NO3–;
щелочь NaOH ![]() Na++ОН–).
Na++ОН–).
Наличие paзнозаряженных ионов объясняет
электропроводность водных растворов электролитов. Различают слабые и сильные
электролиты. Даже вода, хотя и в небольшой степени, является электролитом Н2О
![]() Н++ОН–.
Недиссоциированные молекулы воды являются полярными молекулами – диполями. В результате взаимодействия
катионов и анионов с полярными молекулами воды образуются гидратированные ионы. Процесс гидратации сопровождается выделением
энергии. При соприкосновении металлической поверхности с раствором электролита
происходит взаимодействие между электрически заряженными частицами раствора
(катионами, анионами, гидратированными ионами) и ион-атомами металла,
находящимися на поверхности, приводящее к переходу последних в раствор в виде
гидратированных ионов. Источником энергии, необходимой для разрыва связи между
ион-атомом и электроном является процесс гидратации.
Н++ОН–.
Недиссоциированные молекулы воды являются полярными молекулами – диполями. В результате взаимодействия
катионов и анионов с полярными молекулами воды образуются гидратированные ионы. Процесс гидратации сопровождается выделением
энергии. При соприкосновении металлической поверхности с раствором электролита
происходит взаимодействие между электрически заряженными частицами раствора
(катионами, анионами, гидратированными ионами) и ион-атомами металла,
находящимися на поверхности, приводящее к переходу последних в раствор в виде
гидратированных ионов. Источником энергии, необходимой для разрыва связи между
ион-атомом и электроном является процесс гидратации.
Когда ион-атом металла переходит в
раствор, на поверхности металла остается эквивалентное количество электронов,
которые в раствор не переходят и сообщают участку металла локальный отрицательный
заряд (излишек электронов). Поверхность такого участка металла становится
электрохимически неоднородной.
Таким образом, основное отличие электрохимической
коррозии от химической состоит в том, что коррозионный процесс возникает в
результате протекания тока во множестве коротко замкнутых гальванических
элементах, образующихся из-за неоднородности отдельных участков металла. При
этом одновременно протекают два электродных процесса [16]:
1. Анодный
процесс – переход металлических ион-атомов в раствор в виде катионов с
оставлением соответствующего количества избыточных электронов на поверхности
металла.
2. Катодный
процесс – ассимиляция избыточных электронов ионами или гидратированными
молекулами в электролите (деполяризаторами), которые при этом восстанавливаются
(рис. 12.1) .
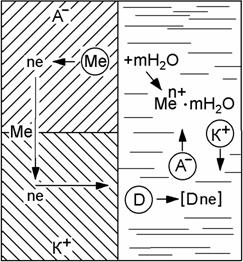
Рис. 12.1 – Схема электрохимического процесса коррозии
Без протекания второго процесса первый
(растворение металла) должен был бы скоро прекратиться.
Наличие электронной проводимости у
металла и ионной проводимости у растворов электролита позволяет анодным и
катодным процессам протекать раздельно на различных участках поверхности
металла. Участок металла, который растворяется, называется анодом, а участок, на котором
имеет место разряд избыточных электронов – катодом. При протекании этих двух процессов имеет место перетекание
электронов в металле от анодных участков к катодным, и соответствующее перемещение
катионов и анионов в растворе.
Таким образом, металл при взаимодействии
с электролитом заряжается отрицательно или положительно, приобретая
определенный электрохимический потенциал.
Это позволяет оценивать устойчивость металла к данной среде по величине и
знаку электрохимического нормального (стандартного) потенциала. [8]
Стали и сплавы, взаимодействующие с
электролитами, можно рассматривать как многоэлектродные элементы, состоящие из
большого числа расположенных на поверхности микрогальванопар. Это объясняется
различными свойствами микро- и макроструктуры, наличием неметаллических
включений, разного уровня дефектности кристаллической решетки и т.д.
Имеются также и другие причины возникновения
локальных коррозионных пар:
1) наличие на поверхности металла
несплошных продуктов коррозии (ржавчины, окалины) или несплошных защитных
металлических покрытий (хром, никель, цинк);
2) неоднородность напряженного и
структурного состояния участков металла: более напряженные и деформированные
участки металла (местной наклеп, изгиб, околошовная зона) являются анодными и
растворяются быстрее;
3) контакт пар металлов с различным
электрохимическим потенциалом (сталь+медь, сталь+цинк, сварной шов+участок
зоны термического влияния и т.п.).
На рис. 12.2 показаны схемы примеров возникновения
коррозионных элементов, а на рис. 12.3 представлена схема коррозии сварного
соединения.
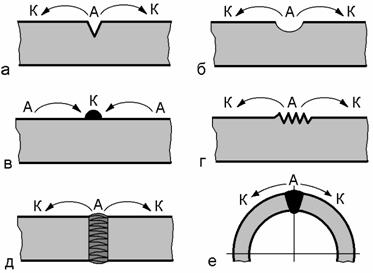
Рис. 12.2 – Схема образования коррозионных элементов:
а – царапина; б – вмятина; в – окалина;
г – местный наклеп; д, е – сварные швы;
А – анод; К - катод
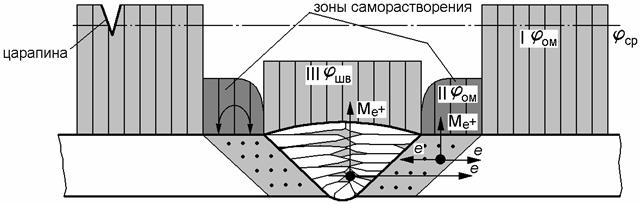
Рис. 12.3 – Схема коррозии сварного соединения
Одним из важнейших факторов коррозии в
водных растворах кислот, щелочей и технической воды является водородный
показатель рН, т.е. логарифм концентрации ионов водорода в растворе, взятый с
обратным знаком.
рН = –Lg Сh,
где Сh –
концентрация ионов водорода.
Величина рН определяет кислый (рН <
7), нейтральный (рН = 7) или щелочной (рН > 7) характер среды и влияет на
скорости коррозии металла в той или иной среде (рис. 12.4) .
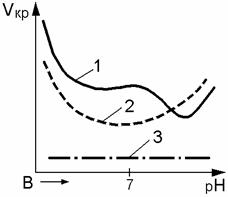
Рис. 12.4 – Типичные кривые скорости растворения
металлов
в зависимости от рН раствора:
1 – железо; 2 – алюминий; 3 – титан
Согласно ГОСТ 5272-66 коррозию разделяют
на следующие виды:
1) атмосферная коррозия – в атмосфере
воздуха;
2) подземная коррозия – в почвах и
грунтах;
3) коррозия блуждающим током, возникающая
от внешнего источника;
4) контактная коррозия –
электрохимическая коррозия, вызванная контактом элементов или участков с
разными потенциалами;
5) биокоррозия – под влиянием
жизнедеятельности микроорганизмов;
6) коррозия при полном погружении – когда
металл полностью погружен в жидкую среду;
7) коррозия при неполном погружении;
8) щелевая коррозия – коррозия в узких
зазорах и щелях между двумя металлами, или участками металла;
9) коррозионное растрескивание – при
одновременном воздействии среды и растягивающих напряжений;
10) газовая коррозия – химическая
коррозия при высоких температурах в среде газов;
11) морская коррозия – в среде морской
воды.
Атмосферная
коррозия приносит наибольший урон:
80 % всего используемого металлофода и 50 % всех потерь металла является
результатом атмосферной коррозии. Атмосферная коррозия в зависимости от степени
увлажнения поверхности металла разделяется на сухую, влажную и мокрую (рис. 12.5) .
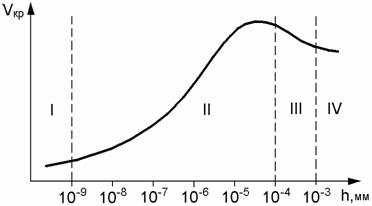
Рис. 12.5 – Виды атмосферной коррозии:
I – сухая; II – влажная; III – мокрая; IV – с полным
погружением
Интенсивность атмосферной коррозии
зависит от количества кислорода, поступающего к корродируемой поверхности. Поэтому
очень тонкие пленки атмосферной влаги опаснее (высокое насыщение кислородом).
Усиливает агрессивность атмосферы ее загрязнение коррозионно-активными агентами
(H2S, NH3, MgCl2). Особо обращают внимание на
местные избирательные виды коррозии сварных соединений из хромистых и
хромоникелевых сталей – межкристаллитную (МКК) и коррозию под напряжением.
Известно [8]
три механизма возникновения МКК в сварных соединениях:
1) обеднение границ зерен хромом за счет
образования равновесных для данных температурно-временных условий карбидных
или карбонитридных фаз (Сr23С6, Сr7С3
при Т = 450...900 0С и TiC, NbC при Т = 1200...1250 0С);
2) образование карбида железа Fe3C по
границам зерен и снижение их cтойкости к агрессивной среде;
3) сегрегация поверхностно-активных элементов
в заданных температурных условиях (S, P, Si, Pb) по границам зерен, снижающая
их стойкость к среде.
По первому механизму МКК развивается в
сталях и сплавах аустенитного классов, а по третьему и частично первому – в
сталях мартенситно-ферритного, мартенситного и ферритного классов.
Температурно-временная область возникновения МКК представлена на рис. 12.6.
Следует отметить, что Ni, Si и Со, повышая термодинамическую активность
углерода, способствуют увеличению количества карбидов и понижают значение tmin времени сохранения стойкости стали к агрессивной
среде. В то же время Mn, Mo, W, V, Nb, Ti повышают стойкость стали и сварного
соединения против МКК.
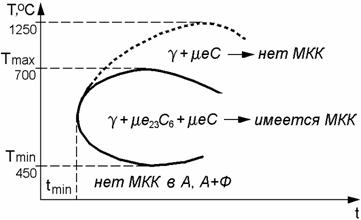
Рис. 12.6 – Температурно-временная область склонности
к МКК
Коррозия
под напряжением сварных соединений
возникает вследствие:
·
сообщения
металлу добавочной энергии, в связи с чем иону Me+, находящемуся на
поверхности, легче покинуть кристаллическую решетку деформированного металла
по сравнению с недеформированным;
·
нарушения
сплошности и защитных свойств поверхностных пленок под действием деформации;
·
повышения
степени неоднородности и появления новых анодных участков при выходе на
поверхность дефектов кристаллического строения (дислокаций, вакансий).
Для сварных соединений чрезвычайно
опасными являются межкристаллитная (в частности, ножевая) коррозия и коррозионное
растрескивание (ГОСТ 5272-76).
Для сравнения скоростей общей коррозии
используют шкалу коррозионной стойкости (табл. 12.1).
Таблица 12.1 – Шкала коррозионной стойкости.
|
Группа
стойкости |
Скорость
коррозии, мм/год |
Балл
стойкости |
|
Совершенно
стойкие |
0,001 |
1 |
|
Весьма
стойкие |
0,001…0,005 0,005…0,01 |
2 3 |
|
Стойкие |
0,01…0,05 0,05…0,1 |
4 5 |
|
Пониженно-стойкие |
0,1…0,5 0,5…1,0 |
6 7 |
|
Малостойкие |
1,0…5,0 5,0…10,0 |
8 9 |
|
Нестойкие |
10,0 |
10 |
Особенности,
и механизмы коррозионных разрушений сварных соединений, зависят от
физико-химического воздействия процесса сварки, вызывающего появление различного
вида неоднородностей, неблагоприятное изменение структуры и свойств металла и
напряженного состояния.
Как
видно из рис. 12.3, сварное соединение в
коррозионном отношении представляет собой сложную многоэлектродную
короткозамкнутую электрохимическую систему, характерными макроэлектродами
которой являются шов, зона термического влияния (с серией переходных структур)
и основной металл.
Процесс
коррозионного растрескивания сварных соединений при статических и динамических
нагрузках в кислых средах протекает быстрее, чем в нейтральных и щелочных
средах и состоит из двух основных этапов:
·
зарождения трещины и инкубационного периода;
·
развития трещины, складывающейся из времени докритического
роста и последующего лавинообразного разрушения.
При
усталостном нагружении скорость разрушения поверхности сварного соединения в
присутствии коррозионной среды резко возрастает.
Для
большинства сочетания металлов и контактируемых сред имеются определенные пороговые
значения напряжений sпор, ниже
которых растрескивание не происходит вообще или на определенной базе испытаний.
Как правило, sпор изменяются
в интервале (0,2...1,0)![]() Т [16], а характер зависимости их от
времени показан на рис. 12.7. Установлено, что, чем выше уровень напряженности
сварных соединений (
Т [16], а характер зависимости их от
времени показан на рис. 12.7. Установлено, что, чем выше уровень напряженности
сварных соединений (![]() ост +
ост + ![]() вн.н), тем сильнее сказывается влияние
концентраторов напряжений на численное значение sпор пороговых напряжений.
вн.н), тем сильнее сказывается влияние
концентраторов напряжений на численное значение sпор пороговых напряжений.
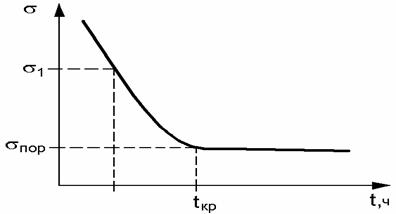
Рис. 12.7 –
Зависимость разрушающих напряжений от времени испытаний
Методы оценки сопротивляемости сварных
соединений
коррозионному разрушению классифицируются [11,8,16]:
1)
по целевому назначению;
2)
по типу объекта;
3)
по типу сред;
4)
по виду напряженного состояния;
5)
по показателям сопротивляемости разрушению.
По
всем перечисленным методам сравнивают результаты испытаний сварного элемента
(шов, зона термовлияния) с основным металлом и изменения того или иного
показателя оценивают относительными коэффициентами, показывающими соответственно
изменение свойств металла шва Кмш или сварного соединения Кс
за время испытания в среде относительно основного металла Ком.